Who spit in the ChIP-seq?
A while back in my previous lab, my officemate performed a ChIP experiment using fruit fly embryos. He was disappointed and surprised to see very little signal and started searching for where the experiment went wrong. There were 95 million reads from the sequencer but when he looked at the reads that matched melanogaster, there were only 4 million! My friend took a handful of the contaminate reads and searched with BLAST to find that a good chunk came from a human genome. Somewhere along the line, a whole bunch of human DNA got into his experiment. Being a careful scientist he wanted to know if he made a mistake and contaminated his data so he gave me the reads and asked if I could figure out where the human DNA came from.
The first question I asked was how much of the contaminating DNA is human. 63.5 million reads matched the human reference genome resulting in information on 701 Mb of the genome and only 0.1% of the genome covered with more than 15 reads. This level of sequencing is not enough to clearly answer an important follow up question: did one person contaminate the sample or many? An approximation can be made by using the mitochondria was sequenced to 1500x (off the charts below). Looking at the mitochondrial variants, that I’m not revealing for privacy reasons, showed that there was only one haplotype. This doesn’t really rule more than one person out but it supports a single contaminator.
Male or Female?
If my colleague contaminated the sample, then there should be Y chromosome DNA and half as much X chromosome DNA compared to the autosomes. The Y chromosome is very degenerated and full of repetitive DNA so we need to filter out all reads that also map to other chromosomes. Once those are thrown out, there are 7454 reads that mapped the the 59Mb Y chromosome. Visual inspection of these remaining reads shows that they often have insertions/deletions in the mapping and are suspect.
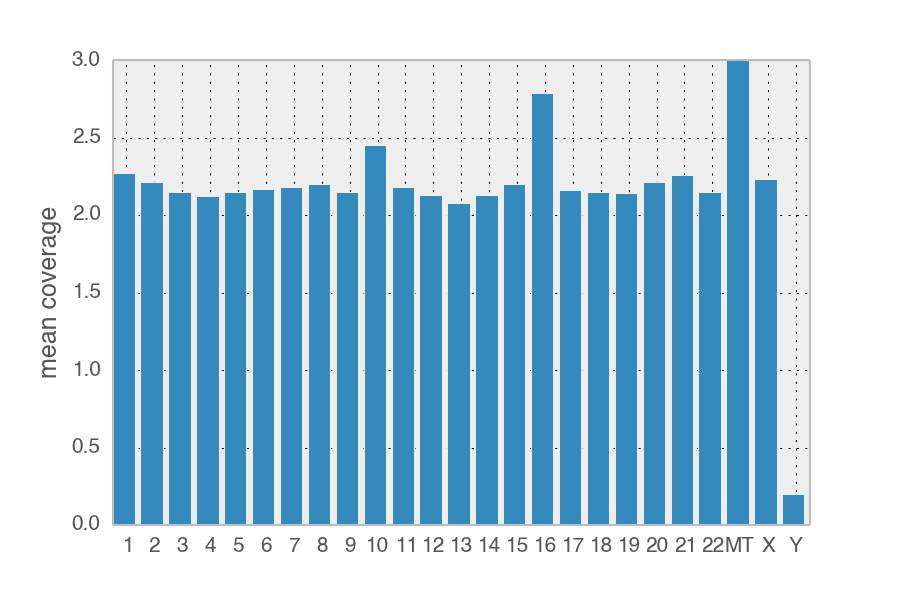
The relative coverage of the X chromosome is with same range of the autosomes, suggesting whoever contaminated the sample is XX and not XY.
Not my officemate but who?
The next person to process the sample was our extremely proficient technician who is normally beyond suspicion but is female along with the rest of the people who process the sample at our sequencing core. They all fit the genetic profile so far which meant I needed to dig a bit deeper. To do this I used the Human Genetic Diversity Project, a collection of polymorphism collected from specific populations across the world. Each population was genotyped at 600,000 positions and I looked at the polymorphisms in the contaminate to see which population it matched the best. After I did this by hand, a great tool called LASER has been released to make this a one step analysis. Plotting the Unknown along with the HGDP samples shows that it clusters nicely in the Central/South Asian group, ruling out our tech and maintaining her spotless record.
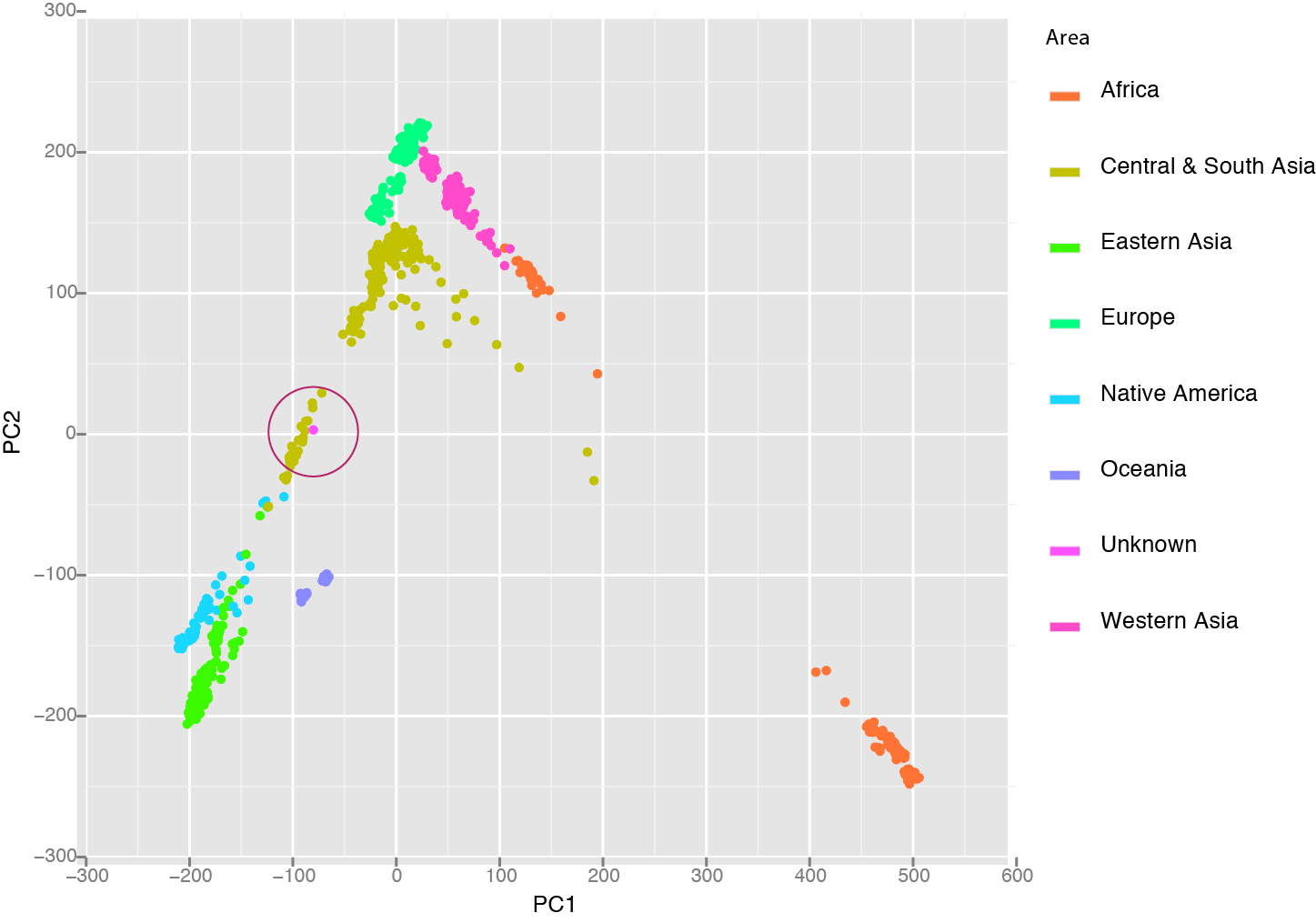
I could show a whole lot more information about this person but won’t because I don’t know who it is and I don’t have consent. I guess I could try to do a facial reconstruction. I purposely did not look at any health related SNPs. I was even a bit worried that the ratio of X/Y/autosome could reveal something invasive like Klinefelter syndrome. At the end of the day, I really wouldn’t be able to say much about this person’s health risk but I can say quite about about her ancestry and so could anyone else who got their hands on a DNA sample.